For over 15 years, Biognosys has been driving progress and innovation in proteomics. Our proprietary next-generation proteomics technology is built on solid science, with over 3,000 publications featuring Biognosys’ workflows, software, and kits. In addition, multiple patents and exclusive licenses protect our unique technology and workflows.
At the core of our cutting-edge proteomics solutions lies our commitment to scientific excellence. We provide multi-dimensional insights on protein expression, function, and structure with industry-leading speed and scalability, catering to a wide range of biological species and sample types.
Our proprietary discovery proteomics technology empowers unprecedented proteome coverage and throughput, allowing for the simultaneous quantification of thousands of proteins across thousands of samples.
Our targeted proteomics technology provides highly sensitive and precise absolute quantification of custom protein panels, relevant to a wide range of species and therapeutic areas.
Our patented technology for the identification of protein structural changes directly in their complex biological environment enables drug target deconvolution and mapping of compound binding sites.
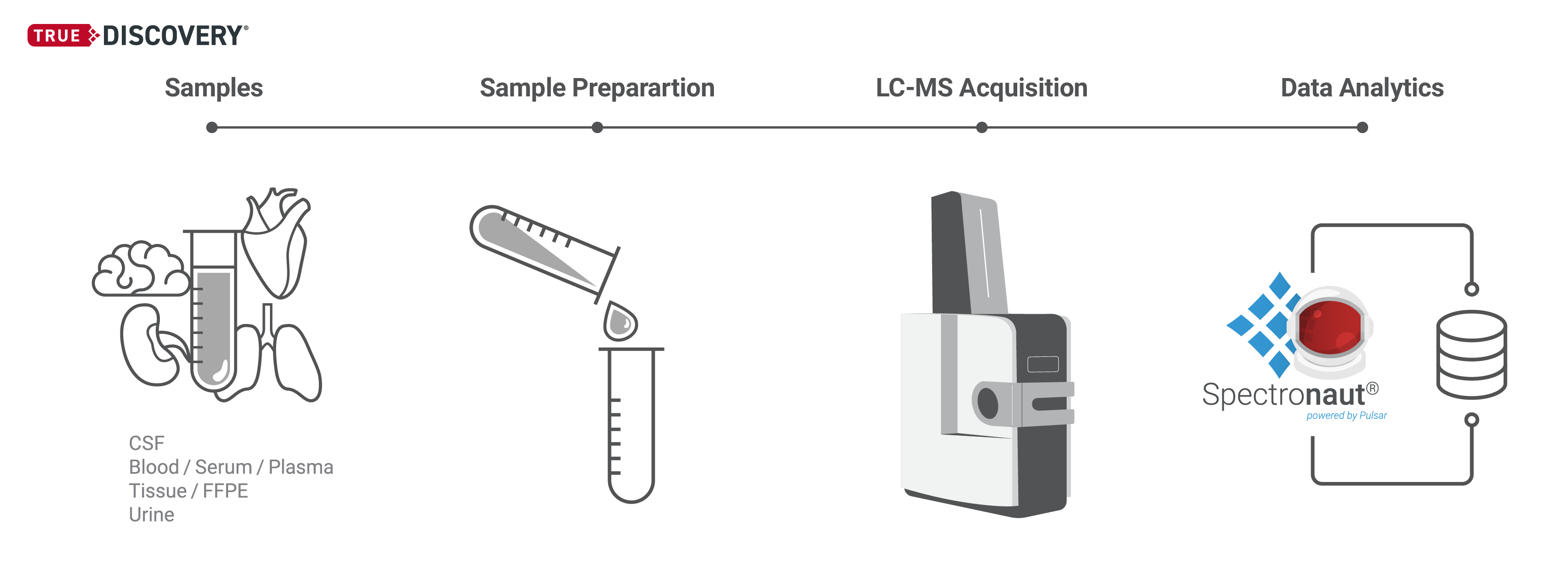
Our TrueDiscovery® platform empowers the quantification of entire proteomes, harnessing our proprietary, patented Hyper Reaction Monitoring (HRM™) technology.
HRM is a Data Independent Acquisition (DIA) based protein quantification workflow developed by Biognosys, delivering unparallel proteome coverage with reproducible and precise quantification of thousands of proteins per sample. The workflow is label-free, ensuring the reliable relative quantification of proteins across multiple biological samples without the need for chemical labeling. The HRM workflow is ideal for identifying differentially expressed proteins in different species and sample types.
Within the HRM workflow, MS data are acquired in DIA mode in a highly parallel process, which provides a comprehensive, peptide-level measurement of all detectable proteins in the sample, with remarkable reproducibility.
The analysis is performed by the most advanced algorithms employed in our flagship Spectronaut® DIA software, including the in-house search algorithm, Pulsar, and advanced Artificial Intelligence (AI) and Machine Learning (ML) signal detection and data normalization based on our patented Indexed Retention Time (iRT) technology. The HRM approach is also extendable to post-translational modifications.
Check out our suite of TrueDiscovery® research services, supporting diverse applications for biomarker discovery, mechanism of action studies, immunopeptidome profiling, and phosphoproteome profiling.
Access our posters and publications on DIA in the Resources section.
PRM and SureQuant are high-resolution mass spectrometry-based targeted data acquisition methods, which enable the precise, multiplexed, and high-throughput quantification of peptide analytes. Biognosys harnesses the unparalleled specificity of these technologies to create customizable assays for monitoring proteins and proteoforms, such as mutations, cleavage products, or isoforms. Additionally, stable isotope-labeled standard (SIS) peptides are introduced as spike-ins, facilitating the monitoring of endogenous analytes and enabling absolute quantification.
Our TrueSignature® contract research services platform provides customizable proteomics panels for pharmacodynamic readouts and clinical biomarker monitoring with exceptional precision.
These panels offer versatile protein combination options, including single proteins, multiplex, and hyperplex panels.
The data analysis within our TrueSignature contract research services is efficiently handled through our proprietary targeted proteomics software, SpectroDive™. SpectroDive is also available as a standalone solution for in-house proteomics, supporting major targeted acquisition methods such as MRM, PRM, and SureQuant.
Access our conference posters on PRM in the Resources section.

LiP-MS is a chemoproteomics approach that enables unbiased profiling of structural protein changes across the entire proteome with a depth of coverage of over 9,000 proteins in human cells. Changes in structural or surface accessibility can modify proteolytic cleavage kinetics during the LiP reaction, leading to distinctive peptide signatures that can be probed using quantitative proteomics approaches such as HRM.
Biognosys applies the LiP-MS technology for drug target deconvolution, identifying and analyzing structure-specific proteolytic states induced by drug binding in cellular lysates in a label-free manner without prior drug modifications. Moreover, we leverage high-resolution limited proteolysis (HR-LiP), a targeted LiP-MS approach, to validate a known target. The target protein can have a high molecular mass or membrane localization and be analyzed at full length in a near-native environment.
LiP-MS was invented by Biognosys’ scientific advisor Prof. Paola Picotti at ETH Zurich. We have deployed our extensive mass spectrometry expertise and machine learning scoring algorithms to develop Biognosys’ proprietary LiP-MS technology and expand its drug target deconvolution ability beyond target identification.
The LiP-MS applications, available as TrueTarget® contract research services, include target ranking, prediction of binding sites, and proteome-wide discovery of induced structural changes. Biognosys is the sole, exclusive provider of proteomics solutions based on LiP-MS technology.
Access key posters and publications on LiP-MS in the Resources section.
Achieve unparalleled precision, depth, and throughput when analyzing protein expression, whether in relative or absolute terms. Utilize our contract research services or leverage our software and kits in-house.
Gain actionable insights on post-translational modifications such as protein phosphorylation via our contract research services or in-house proteomics software and kits.
Uncover comprehensive structural protein changes throughout the entire proteome with our unbiased Drug Target Deconvolution and Validation contract research services.

Biognosys’ innovative technology is featured in over 3,000 publications, many of which are co-authored by our scientific experts in high-impact, peer-reviewed journals..
Access our key technology publications in the Resources section.
Biognosys AG
Wagistrasse 21
8952 Schlieren
Switzerland
Phone: +41 44 738 20 40
US Offices
Biognosys Inc
70 Bridge St, Suite 104
Newton, MA 02458
United States

