Mass spectrometry-based structural proteomics provides deep insights into drug-protein interactions for drug target identification and validation to de-risk your drug development pipeline.
Drug discovery is built on the fundamental principle of pharmacology: find a drug that binds to a specific molecular target and exerts the desired effect within the body to treat or prevent disease. While this may sound simple on paper, it is much more difficult in reality.
A significant number of drugs fail in clinical trials due to poor efficacy and unacceptable toxicity (Fogel, 2018; Sun et al., 2022). All too often, this is because the drug is not binding to the expected target, has significant off-target binding effects, or isn’t working in the predicted way.
Drug target identification and validation are therefore crucial steps in the journey from hit to lead, highlighting the need to fully characterize on-and off-target binding throughout the cell to ensure that a drug is binding where expected and behaving as it should. Once the target is validated and you know where and how a compound binds, you can then optimize it to further improve safety and efficacy.
In turn, this detailed knowledge of drug-target interactions helps to improve the chances of success in clinical trials down the road and de-risk the drug development journey.
Current Drug Target Validation Methods Are Limited
Current techniques for interrogating drug-target interactions have significant constraints that limit their utility for target identification and validation.
For example, X-ray crystallography is time-consuming and technically demanding. It relies on the drug-target complex crystallizing properly and can’t reveal off-target binding elsewhere in the proteome. Cellular thermal shift assays (CETSA) are gaining interest for their ability to probe drug-target interactions within live cells and tissues but require careful optimization for each target (Seashore-Ludlow et al., 2020).
Other techniques, such as affinity-based assays, rely on compound or protein labeling, which can interfere with drug binding. More broadly, many of these established methods do not work well with certain kinds of proteins that could be valuable drug targets due to their large size or inaccessible subcellular location, such as in the cell membrane.
Limited Proteolysis Coupled with Mass Spectrometry: A New Era of Structural Proteomics Technology
Recent technical advances in structural mass spectrometry proteomics now offer an alternative option: limited proteolysis mass spectrometry (LiP-MS).
This technology has been co-developed by Biognosys and the group of Professor Paola Picotti at ETH Zurich, and is the foundation for Biognosys’ TrueTarget™ research platform. TrueTarget offers a simple way of studying drug-target interactions at scale without the need for fiddly optimization, crystallization, modification, or labeling.
Drug-treated and control cell extracts are digested with proteases, creating distinctive repertoires of peptides depending on where and how the drug is bound. These peptides are identified and quantified with high-precision mass spectrometry to generate unique ‘fingerprints’ of drug binding events (figure 1).
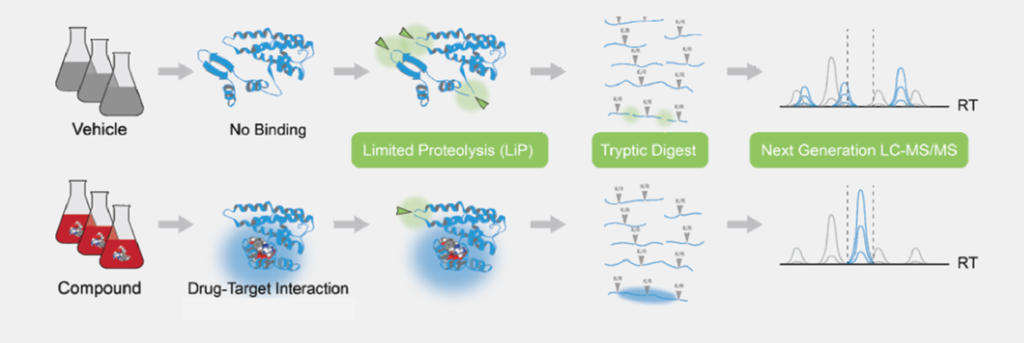
Figure 1: The TrueDiscovery™ LiP-MS workflow
The TrueTarget platform offers two main applications for drug target identification and validation:
• The Drug Target Deconvolution service leverages quantitative LiP-MS for drug target identification and prediction of likely toxicities by revealing all on- and off-target binding events within the cell. Based on a proprietary machine-learning algorithm developed by Biognosys, the service provides a ranked list of predicted targets across the entire proteome.
• The Drug Target Validation service utilizes high-resolution LiP-MS (HR-LiP) to take a closer look at specific interactions, enabling peptide-level resolution of exactly where the drug is binding to the target. This data provides deep structural and mechanistic insights to support target validation and lead optimization.
Applying LiP-MS for Drug Target Identification
Our quantitative whole-proteome LiP-MS approach (also known as LiP-Quant) can be used to map potential drug binding sites and estimate relative binding affinities across the whole proteome (Piazza et al., 2020).
The value of LiP-MS was shown in a study recently published in ACS Chemical Biology (Hendricks et al., 2022) and presented at the EORTC-NCI-AACR (ENA) Symposium, exploring the targets of a novel inhibitor of CDK9 – a transcriptional regulator that plays a key role in cell cycle progression and is an important drug target in cancer.
There are more than 20 CDKs in the human proteome, many of them very similar. Despite plenty of interest from pharma companies, CDK inhibitors have had mixed results in clinical trials, likely due to lack of specificity.
Working in collaboration with AstraZeneca and Paola Picotti’s team at ETH Zurich, we compared four different methods to explore inhibitor binding: conventional affinity chemoproteomics, Kinobeads assay, Cellular Thermal Shift Assay (CETSA), and proteome-wide LiP-MS.
All four techniques homed in on CDK9 as the highest affinity target of the drug, with the LiP-MS analysis showing additional high-affinity binding to CDK11A and CDK16 and revealing the likely binding sites within these proteins. While each technique has its pros and cons, they provide a valuable picture of on- and off-target binding across the entire proteome to support lead compound selection.
Watch our video to learn more:
Using HR-LiP to Characterize Drug Binding Sites for Target Validation
While LiP-MS can be used to characterize drug-target interactions across the entire proteome, high-resolution LiP (HR-LiP) provides a structural look at the interactions between a specific target and drug. This enables characterization of the binding site with peptide-level resolution and provides insights into the drug’s mechanism of action, such as conformational changes or allosteric effects.
In collaborative studies with researchers at the University of Cambridge and Cedilla Therapeutics, presented at the 2022 ENA meeting, the binding sites of two important oncology drugs with hard-to-characterize binding sites were accurately mapped: the EGFR inhibitor gefitinib and the BRD4 inhibitor JQ1. The analysis revealed new insights into the inhibitors’ binding sites and mechanisms of action that were not detectable with other methods.
In another study published recently in Nature Communications (Wrobel et al., 2022), researchers from the University of Cambridge , AstraZeneca, and Biognosys used HR-LiP to map the binding site of a novel small molecule activator of Valosin-containing protein (VCP) D1 ATPase activity – a potential drug target in neurodegenerative disorders such as Huntington’s disease (figure 2).
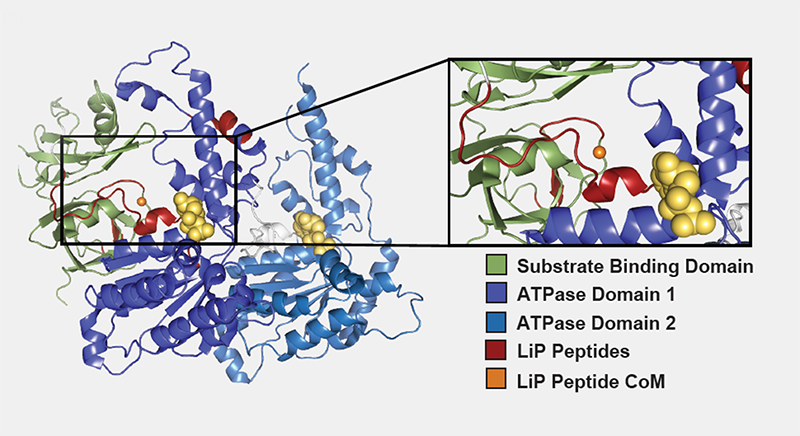
Figure 2: HR-LiP Predicts Activation Site of VCP. Center of mass (orange dot) of LiP peptides (red) indicates binding in cleft between substrate binding domain (green) and ATPase domain 1 (purple), a previously demonstrated activation site for VCP.
TrueTarget: a Unique Platform for Understanding Drug-target Interactions
Drug Target Deconvolution and Validation are exclusively available as research services through Biognosys’ TrueTarget™ platform – a powerful addition to the drug discovery and development toolbox to probe drug-target interactions across the whole proteome.
Our high-throughput pipeline works on thousands of proteins in their native state anywhere within the cell and in any species, providing easy transferability between preclinical models. For proteome-wide LiP-MS, all we need are samples of treated and untreated cell lysate, while HR-LiP requires a cell line overexpressing your target of interest and a stock of your compound.
Get in touch to discover how TrueTarget™ can accelerate and de-risk your drug development pipeline.
References:
- Fogel DB. Factors associated with clinical trials that fail and opportunities for improving the likelihood of success: A review. Contemp Clin Trials Commun. 2018, Aug 7;11:156-164. doi: 10.1016/j.conctc.2018.08.001
- Sun, D et al. Why 90% of clinical drug development fails and how to improve it? Acta Pharmaceutica Sinica B. 2022, 12(7). doi:10.1016/j.apsb.2022.02.002
- Seashore-Ludlow B, Axelsson H, Lundbäck T. Perspective on CETSA Literature: Toward More Quantitative Data Interpretation. SLAS Discov. 2020 ,Feb;25(2):118-126. doi: 10.1177/2472555219884524
- Piazza I, et al. A machine learning-based chemoproteomic approach to identify drug targets and binding sites in complex proteomes. Nat Commun. 2020 Aug 21;11(1):4200. doi: 10.1038/s41467-020-18071-x
- Hendricks JA, et al. Mechanistic Insights into a CDK9 Inhibitor Via Orthogonal Proteomics Methods. ACS Chem Biol. 2022, Jan 21;17(1):54-67. doi: 10.1021/acschembio.1c00488
- Wrobel L, et al. Compounds activating VCP D1 ATPase enhance both autophagic and proteasomal neurotoxic protein clearance. Nat Commun. 2022, Jul 16;13(1):4146. doi: 10.1038/s41467-022-31905-0

