SpectroMine 3 – our powerful and user-friendly solution for DDA proteomics – comes with significantly increased processing speed and new functionalities for specialized workflows such as PTM analysis and immunopeptidomics. Take advantage of the most powerful search engine for isobaric labeling quantification with support for the latest acquisition methods and labeling reagents.
“SpectroMine is a game-changer for the analysis of multiplexed proteomics experiments. The speed and quality of analysis are unsurpassed.”
Dr. Mikhail Savitski, EMBL Heidelberg

Analyzing biological samples with complex post-translational modification (PTM) profiles is important to understand many biological processes. However, the detection of additional PTMs can have a severe impact on processing time. SpectroMine 3 is designed to handle such large search spaces efficiently, resulting in impressive speed improvements over previous versions.
The concurrent profiling of multiple PTMs across multiple experimental conditions is particularly computationally expensive. Adding modifications increases the complexity of the search space exponentially, leading to significantly increased processing time with most search engines, potentially resulting in a productivity bottleneck. SpectroMine 3 is optimized for fast search of such datasets while maintaining excellent identification performance.
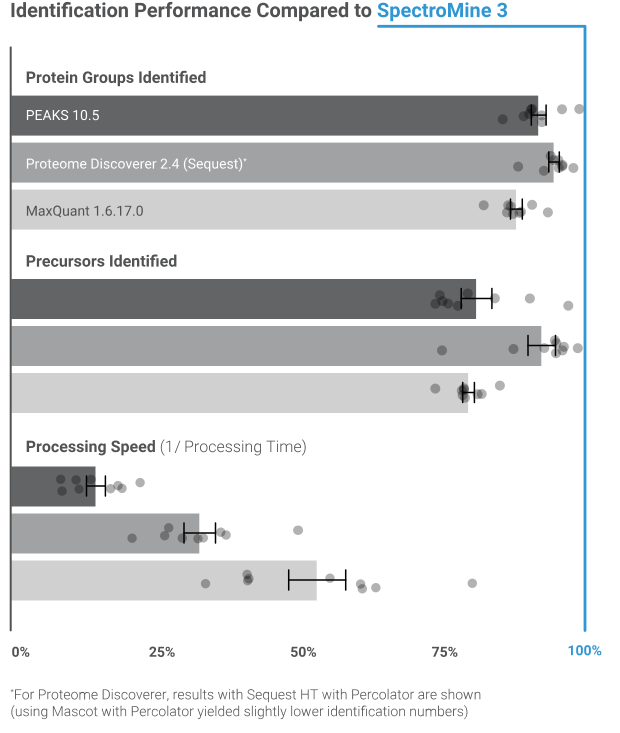
Our example shows key metrics from the analysis of a published dataset of enriched phospho peptides from a human cell culture experiment with the SARS-CoV-2 virus1. Up to 5 variable PTMs per peptide were allowed, including phosphorylations, oxidation, and acetylations. SpectroMine outperforms other search engines in processing speed and identified precursors (see Figure 2). As a result of optimized handling of large search spaces, this analysis benefits from a 70% speed increase with the newest SpectroMine version.
SpectroMine brings the power of Pulsar, our industry-leading and vendor-independent database search engine to DDA proteomics for deep proteome coverage and fast analysis.
With advanced deep learning models for indexed retention time (iRT) and fragmentation prediction, SpectroMine achieves unsurpassed proteome coverage. We tested a range of software products using harmonized search space settings, protein inference, and False Discovery Rate (FDR) control. The results for datasets with and without isobaric labeling demonstrate that SpectroMine provides the most identifications and fastest processing times for a wide range of DDA experiments (see Figure on the left).
SpectroMine lets you take full advantage of the latest instrumentation from all major vendors, including FAIMS Pro™ and PASEF™ ion mobility technologies, delivering state-of-the-art identification performance and short processing times.
At the same time, SpectroMine provides an intuitive interface to ensure data integrity and enable the sharing of data. FDR is rigorously controlled at Peptide Spectrum Match (PSM), peptide and protein levels, and a wide range of quality control visualizations are also available. To keep your results organized, they are saved in search archives, giving you full access to annotated raw data, and making it easy to share with anyone through the free SpectroMine Viewer.
SpectroMine provides a user-friendly environment to get DDA proteomics results in just a few steps, with extensive data browsing and result reporting options.
This video is a brief introduction to using SpectroMine for isobaric labeling quantification (ILQ). Follow along as Fabia Simona from our Product Support team demonstrates the most important analysis steps for setting up an ILQ analysis for fractionated samples.
You will also learn about some of the new functionalities in SpectroMine 3, including PTM site-level analysis, fraction summarization, and user interface improvements that make analyzing your DDA data more convenient than ever.

SpectroMine provides a complete platform for isobaric labeling experiments. With integrated analysis templates for all major labeling reagents, staying on top of the latest innovations in the field is made easy.
DDA combined with isobaric labeling quantification (ILQ) is a discovery proteomics workflow, enabling quantification of multiple biological samples simultaneously in a single run (see upper Figure). ILQ delivers highly precise and reproducible quantification with deep proteome coverage [Muntel, 2019].
In just a few steps, ILQ analyses can be set up quickly and efficiently (see Figure on the left). With automated calibration and smart default settings, optimal results can be achieved with minimal user input.
For experiments where the number of samples exceeds reporter multiplexity, samples can be organized in blocks/batches. SpectroMine offers experiment-wide normalization across these blocks. Moreover, workflows for assessing labeling efficiency allow you to quickly correct for sample preparation-related differences between analysis channels.
Quantification results can be viewed directly in SpectroMine, and then exported in either a customizable report format or with pre-packaged schemas for downstream processing with external tools such as MSstatsTMT.
SpectroMine can be configured for all Isobaric Labeling reagents and comes with presets for:
Biognosys AG
Wagistrasse 21
8952 Schlieren
Switzerland
Phone: +41 44 738 20 40
US Offices
Biognosys Inc
70 Bridge St, Suite 104
Newton, MA 02458
United States